
Translate this page into:
Magnetic resonance imaging of brain findings in hyperammonemic encephalopathy
This is an open access article distributed under the terms of the Creative Commons Attribution NonCommercial ShareAlike 3.0 License, which allows others to remix, tweak, and build upon the work non commercially, as long as the author is credited and the new creations are licensed under the identical terms.
This article was originally published by Medknow Publications & Media Pvt Ltd and was migrated to Scientific Scholar after the change of Publisher.
Sir,
Acute hyperammonemic encephalopathy is a fatal condition.[123] Hyperammonemia occurs when ammonia is overproduced or insufficiently cleared and is a cause of encephalopathy.[1] Hepatic failure is the most frequent culprit of hyperammonemia, other etiologies include congenital enzyme deficiencies of urea cycle, drugs such as valproic acid, barbiturates, narcotics, acetaminophen, and chemotherapy, urease-producing gastrointestinal or urinary tract organisms (examples: Proteus, ureaplasma urealyticum, Helicobacter pylori), gastrointestinal bleeding, renal disease, ureterosigmoidostomy, parenteral nutrition, Reye syndrome, severe muscle exertion, and septic shock.[1234] We present a fatal case of acute hyperammonemic encephalopathy secondary to liver failure related to hepatitis B.
Case Presentation
A 69-year-old female patient presented to emergency room with episodes of altered mental status over 24 hours. Her medical history was significant for hepatitis B-related liver cirrhosis/encephalopathy and chronic kidney disease. Patient was started on lactulose and rifaximin for hyperammonemia of 291 µmol/L. Computed tomography scan of head demonstrated no mass, hemorrhage, or stroke. Patient was admitted to the Intensive Care Unit and intubated for acute respiratory failure. She remained unresponsive despite 48 hours of lactulose and rifaximin.
The magnetic resonance imaging (MRI) of the brain without contrast on day 3 showed a dark rim involving the globus pallidi bilaterally on the susceptibility weighted images and central high signal seen within the globus pallidi on the T2-weighted images. In Figure 1, diffusion-weighted imaging (DWI) visualized multiple areas of restricted diffusion involving the lateral portion of the temporal lobes extending up to the posterolateral portions of the parietal lobes along with the medial portions of the frontal lobes, thalami bilaterally, centrally within the midbrain, and also the periaqueductal gray matter.
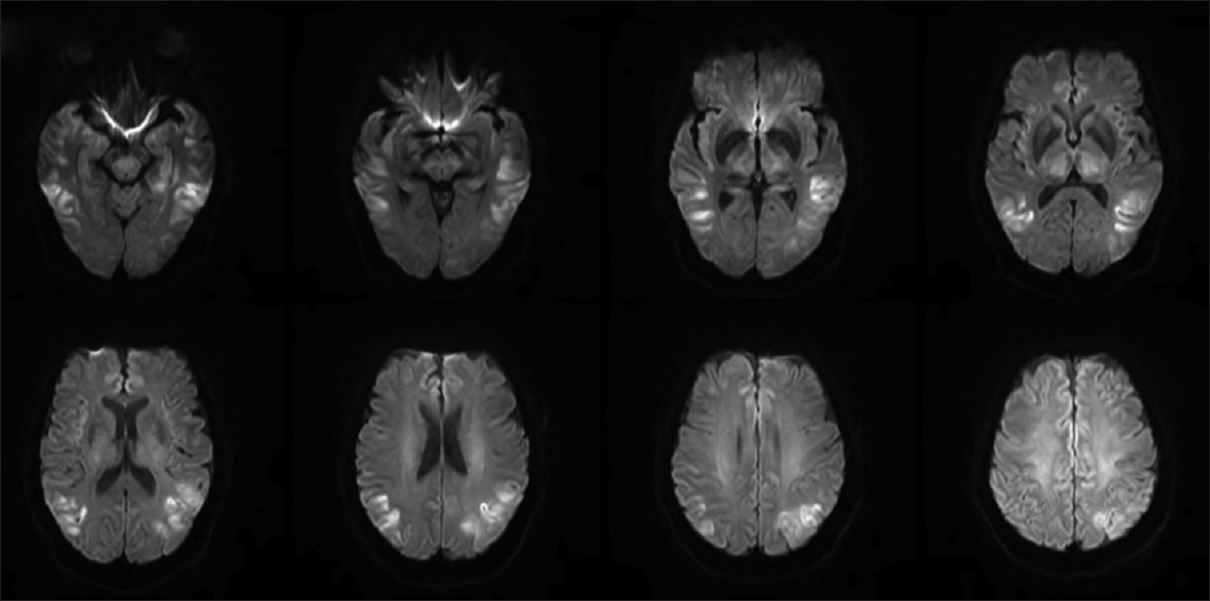
- Diffusion-weighted imaging showed multiple areas of restricted diffusion involving the lateral portion of the temporal lobes extending up to the posterolateral portions of the parietal lobes along with the medial portions of the frontal lobes, thalami bilaterally, centrally within the midbrain, and also the periaqueductal gray matterthe
Repeat MRI on day 6 [Figure 2] showed marked progression of diffuse cortical injury involving both cerebral hemispheres. The injury to the thalami and central structures has partially pseudonormalized on DWI and relative sparing of the perirolandic/motor-sensory cortex and cerebellum. Mild, diffuse cerebral swelling and sulcal effacement from the diffuse cortical injury without midline shift. Due to multiple medical complications, unresponsiveness to medical management, and poor prognosis, patient's family members decided to withdraw the life support. She was terminally extubated and died 9 days after hospitalization.
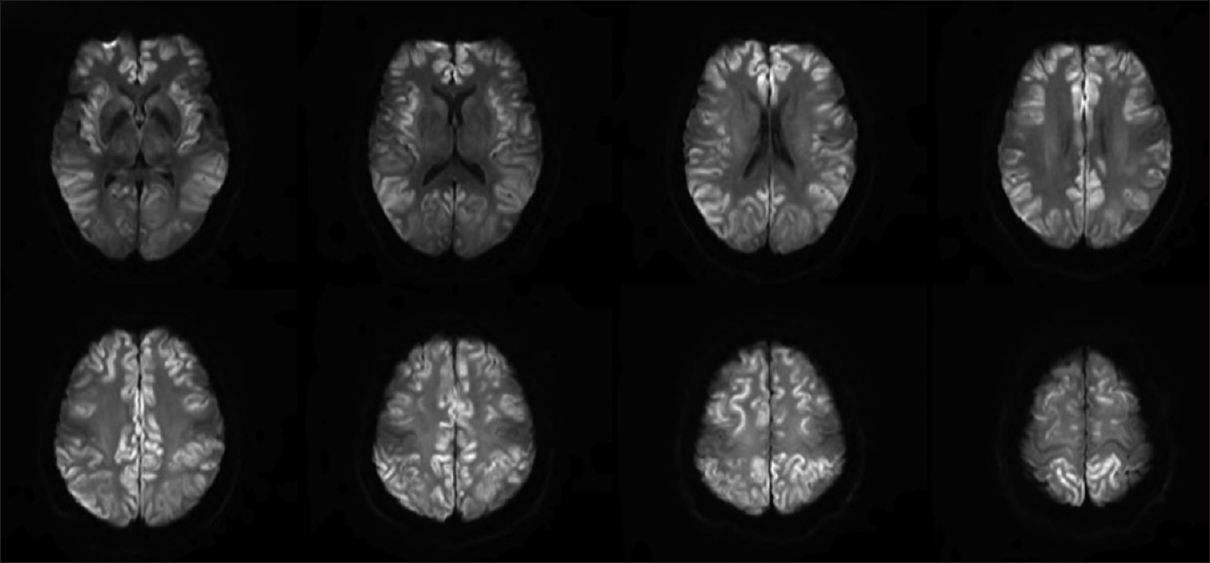
- Repeat magnetic resonance imaging on day 6 showed marked progression of diffuse cortical injury involving both cerebral hemispheres and relative sparing of the perirolandic/motor-sensory cortex and cerebellum. Mild, diffuse cerebral swelling and sulcal effacement from the diffuse cortical injury without midline shift
Discussion
Hyperammonemia is an end result of several metabolic disorders including hepatic encephalopathy and urea cycle disorders such as deficiency of carbamoyl phosphate synthase or ornithine transcarbamylase.[34] In a case series, three patients with acute hyperammonemic encephalopathy were misdiagnosed as hypoxic-ischemic encephalopathy, restricted diffusion on DWI involving the entire cortex, in particular, the insular and cingulate cortices as well as the deep gray matter of the basal ganglia.[1] The first patient had a history of end-stage liver cirrhosis due to alcohol abuse presented with hyperammonemia of 425 µmol/L died 14 days after hospitalization. The second patient had elevated ammonia levels (233 µmol/L) due to alcoholic hepatitis and discharged to skilled nursing facility due to poor prognosis. The third patient had hyperammonemia (217 µmol/L) and hepatic toxicity secondary to acetaminophen and she recovered neurologically in 9 days after admission. In another case report, patient presented with hyperammonemia due to liver disease (111 µmol/L) and MRI revealed peripheral cortical hyperintensity in both temporal lobes and right posterior cingulate gyrus with corresponding diffusion restriction on DWI and T1-weighted hyperintensity in both basal ganglia.[4] Patient was successfully recovered with medical management.[4]
In another report, three patients were found to have acute hyperammonemic encephalopathy due to late-onset ornithine transcarbamylase deficiency.[3] These patients had restricted diffusion particularly involving insular cortex and cingulate gyrus on DWI with sparing of the perirolandic and occipital cortices.[3] The deep gray matter structures, white matter, and cerebellum were normal. Two patients with the maximum blood ammonia level of 235 µmol/L and 293 µmol/L were recovered from acute event successfully, and one patient with ammonia level of 2050 µmol/L died 5 days after admission.[3] Takanashi et al. described the vulnerability of the cingulate gyrus and insular cortex due to hyperammonemic-hyperglutaminergic encephalopathy, with relative sparing of perirolandic and occipital cortex.[3] Similarly, brain MRI showed diffuse bilateral restricted diffusion in nearly the entire cerebral cortex due to hyperammonemia following valproic acid exposure for schizoaffective disorder.[2]
Conclusion
Hyperammonemic encephalopathy should be considered in the appropriate clinical setting without hypoxic event. MRI on DWI shows restricted diffusion involving cerebral cortex and thalamus bilaterally with relative sparing of motor, sensory cortex, and cerebellum. Early diagnosis and aggressive treatment are critical as it can be fatal.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
- Diffusion-weighted imaging in acute hyperammonemic encephalopathy. Neurohospitalist. 2013;3:125-30.
- [Google Scholar]
- Fatal hyperammonemic brain injury from valproic acid exposure. Case Rep Neurol. 2012;4:224-30.
- [Google Scholar]
- Brain MR imaging in acute hyperammonemic encephalopathy arising from late-onset ornithine transcarbamylase deficiency. AJNR Am J Neuroradiol. 2003;24:390-3.
- [Google Scholar]
- MRI findings in acute hyperammonemic encephalopathy resulting from decompensated chronic liver disease. Acta Neurol Belg. 2012;112:221-3.
- [Google Scholar]




